

NCBIにゲノム配列を登録する際のTipsの備忘録。
ゲノムアセンブリーデータとSRA(生リードデータ)のアップ方法を書いておきます。
(NCBIにアノテーションファイルはアップロードしたことない)
ENAへの登録方法も以前書いてます→ENAに配列データを登録する
正直ENAの方が簡単です。
手順
1. 配列準備
- 配列名はシンプルにしておく(ex. Hoge_scaffold0001)
- 200 bp以下の配列は消す
- seqkit seq -m 200
2. NCBIにログイン
NCBIのSubmitのページに行き、ログイン(ORCIDでログインできる)。
3. ゲノムアセンブリー登録の場合
大まかな手順
- アセンブリーにまつわる情報をフォームに従って記入
- 配列をアップロード
- NCBIの自動チェックが進行
- アダプター配列、オルガネラなどのコンタミがあれば、コンタミで検出した結果がメールで通知。
- フォームに書いてあることが変だったり、登録済みの同種のゲノムサイズと大きくズレている等があると、おかしいですよとメールで通知。
- 場合に応じて、コンタミを削除やN置換、または理由説明のメールの返信をする
- 自動チェックで問題ないとStatusが「Genomes: Processing」となり、その後のなんらかのチェックも通過すると「Genomes: Processed」となり、公開日に公開される。
- 自動チェック以降は人がチェックしていると思われる。
Release date(公開予定日)をすごく先にしていると、優先順位が下がるっぽいので、急いでいる人は即時公開にするといいらしい。
- 自動チェック以降は人がチェックしていると思われる。
フォームに入力
- Submission portalの「GenBank」>「Submit assembled prokaryotic and eukaryotic genomes」>「Submit」のページに移動。
- 「New submission」をクリック。
- Submission Typeを選ぶ
- 2倍体ゲノムのHaploidの一つや1倍体ゲノムをアップロードする場合は
とりあえず「Single genome」でよい
- 2倍体ゲノムのHaploidの一つや1倍体ゲノムをアップロードする場合は
- 順番に記入していく
- Continue押しても戻れるのでわかるところから埋めて大丈夫
- 以下それぞれの注意事項
1. SUBMITTER
- このページのSubmitterやGroup for this submissionは投稿作業の進捗が共有されるグループというだけなので、公開ページの「Submitter」とは関係ないらしい
2. GENERAL INFO
- BioProjectやBioSampleが既に取得されていれば(過去の登録と統合したいなど)、
ここで記入 - Release dateは、とりあえず「Release on specified date or upon publication, whichever is first」にしてテキトウな日付にしても大丈夫。あとから変えられる。
- Genome infoのYes/Noは、以下で大丈夫。
- Did your sample include the full genome? - yes
- Is this the final version? – yes
- Is it a de novo assembly? – yes
- Is it an update of existing submission? – no
- 同種のゲノムが既に登録されていたとしても、自分の過去の登録をマイナーチェンジしたい場合以外は、ここはnoでよい
- □Do not automatically trim or remove sequences identified as contamination – チェック入れない
- 配列をアップロードすると、アダプター配列などを勝手にトリミングしてくれる。
※ここ以降はBioProjectやBioSampleを登録するかどうかで、フォームの各セクション番号がずれるので、番号はあまり気にしないでください。
3. FILES
Which of these options describes this genome submission?
の質問が微妙にわかりにくいが、要するにChromosome levelで登録するか、それともScaffoldにするかの分かれ目がこの質問のようです。
1. Each chromosome is in a single sequence and there are no extra sequences
→Chromosome level。この後のASSIGNMENTで配列との対応付けをするが、Scaffold1がChr1で、Scaffold2がChr2でのように、対応付けられるならこっち。
2. One or more chromosomes are still in multiple pieces and/or some sequences are not assembled into chromosomes
→1でないならこっち
Upload
やりやすい方法でいいが、無難にやるならFTP。
4. GAPS
ギャップ(N)があるとここで各長さと数が表示される。
HiC scaffoldingなどをしているなら以下のように選択。
- Did you randomly merge the sequences into a single sequence (for example, maybe you just linked the sequences together by size without using an assembler program)? – No
- Appropriate minimum number of Ns in a row (0-10) that represents a gap – 10
- 最小のギャップの長さ。スキャフォールディングする際に挿入したNの数は大抵100以上とかなので10で良い。
- Do any of the N’s represents gaps of completely unknown size (the gap size was NOT estimated by an assembly program and a single value, eg 100, was used)? – Yes
- Are all gaps of unknown size represented by the same number of N’s, eg 100? – Yes
- Number of N’s in gap of unknown length – 100
- ギャップがスキャフォールディングのソフトによって挿入されているなら、その長さをここで記入。とはいえページ頭のGap lengthの値を入れればよい。
- What type of evidence was used to assert linkage across the assembly gaps? – proximity-ligation
- スキャフォールディングがどんな方法で行なわれたか。
5. ASSIGNMENT
染色体番号やオルガネラを割り振りたい配列がある場合はここで記入。
6. REFERENCES
論文を結びつけたい場合はここで記入。公開後でも変更できる模様。
7. REVIEW & SUBMIT
確認してSubmit。ここからNCBIの自動チェックが進行する。大体半日くらいでチェックが終了する。
以降、コンタミ処理の対応例。
コンタミ処理
アダプター配列などが検出されるとエラーがある旨のメールが届く。
フォームからDetailsをみると以下のようなファイルがダウンロードできる。
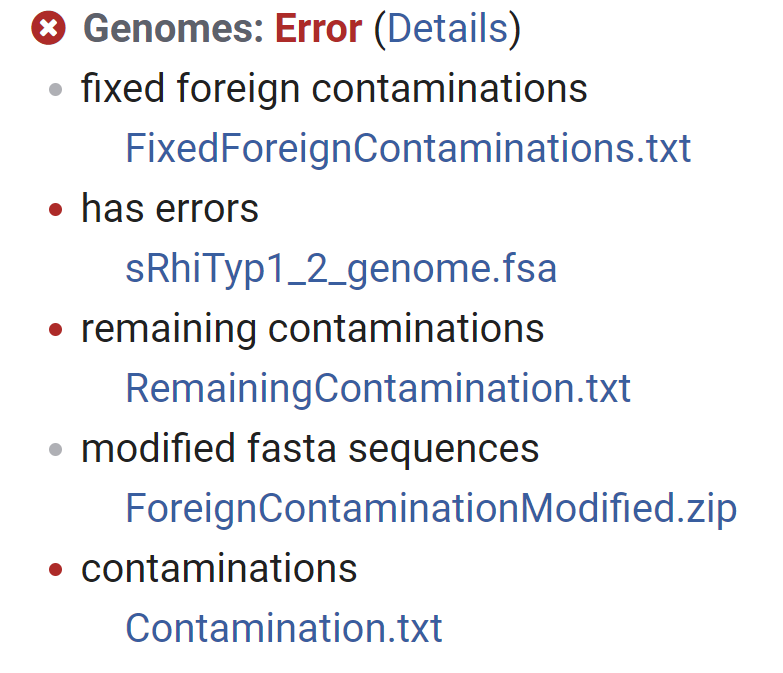
検出された端っこのアダプター領域がFixedForeignContaminations.txtに記述されており、
端だけトリミングされたあとの配列がForeignContaminationModified.zipに入っている。
さらに配列中に見つかったコンタミらしき配列の座標やオルガネラ配列のレポートがRemainingContamination.txtに書かれている。
このテキストファイルの座標は、端がトリミングされたあとのForeignContaminationModified.fastaに基づく(元ファイルじゃない)。
配列ごと消したり、オルガネラを分けたりは適宜する。
さらにコンタミらしき領域を全部Nに変換するなら例えば以下のようにできる(依存:BEDTOOLS)。
コンタミ領域をN変換
awk '/^Trim:/{flag=1; next} /^$/{flag=0} flag' RemainingContamination.txt |\
awk '
BEGIN{FS="\t"; OFS="\t"}
/^Sequence name/ || /^$/ {next}
{
split($3, a, /\.\./);
start = a[1]-1;
end = a[2];
print $1, start, end
}
' > ForN.bed
bedtools maskfasta -fi ForeignContaminationModified.fasta -bed ForN.bed -fo genome_masked.faキレイキレイしたら「Fix」から再アップロード。
SRA(生リードデータ)の登録の場合
Submission portalの「Sequence Read Archive (SRA)」>「Submit」のページに移動。
ほとんど「ゲノムアセンブリー登録の場合」と同じ。
3. SRA METADATA
アップロードするファイル名をfilename, filename2….の列にファイル名を列挙して書けば、同じSRX0000のようなアクセッション番号に格納される。
圧縮ファイルもOK(tar, gzip, bzip2)だけど、zipはだめ。
(備考)Taxon、BioProject、BioSample
Taxon
TaxonIDは各ランクに振られており、その階層関係がhttps://www.ncbi.nlm.nih.gov/guide/taxonomy/に、以下のように記録されている(一部抜粋)。V1が親ID, V3が子ID, V5が子の階層名。
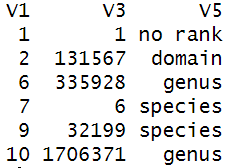
この階層関係が厳しく決まっているため、新たな種の追加は簡単にできるが、階層関係の変更は基本的に受け付けてもらえない(A属を、B科の下じゃなくてC科の下にしたいなどのオーダーは厳しい)。
ゲノム配列の登録の際は、ランクが(基本的には)Speciesのものしか登録できない。Speciesとして登録されていない場合は新しくTaxonの申請が必要。
BioProject
論文一つにつき一つのBioProjectを割り当てている例が多い。
ゲノムはもちろんSRAの登録も、同時に登録するデータは全て一つのBioProjectと紐づくので、
別のBioProjectにしたいならSubmissionのプロセス自体を分ける必要がある。
論文のアブストなどの省略版を「Public description」に書いておくと論文の宣伝にもなる。
公開後、BioProjectのページにはPublicationsも記載される。これは後からも追記できるので、論文がPublishされたあとは絶対追加した方が良いし、ゲノム配列は元論文が引用されないことも多々あるので論文を紐づけておくと引用する側も楽ちん。
BioSample
BioSampleの分ける基準は非常に難解だし、登録されているものもかなり混迷を極めている。
概ね、同じ個体なら同じBioSampleとしてもよいかと思うものの、生物によって個体の単位が難しかったりする。特にRNAデータなどの場合は、BioSampleの登録のごとに細かく組織やステージの情報が付随できるために、非常に細かく登録分けがされている印象がある。
つまり、元も子もないが、BioSampleの分類に一貫性をもたせることを諦めて、論文のサプリなどにまとめた表などを入れるのが親切かと思います。

コメント